
Deep Omics
Analysis Platform
With the Focus on Cancer Studies
This DOAP is a cloud-based environment for conducting bioinformatics research. It is a central hub for research teams to store, analyze, and jointly interpret their omics and relevant data. The platform integrates analysis pipelines, omics data, and visualization tools for researchers to understand their data. Also, the platform provides user-friendly environments for developers to implement bioinformatics tools.
Our Focus
Biomedical Data Analysis at Scale.

Biomedical Data Analysis
Health care professionals, including clinical researchers could use pipelines to analysis data and view reports.

Module Development
Technology or product developers, R&D specialist could develop and pulish analysis module online.

Health App
Anyone could upload their genome data and interpret the information by the mobile application.
Features
Data Collection
Over 200TB data including the RNA-seq and microarray data.
Online File Manager
Online file manager and code editor supported.
Analysis Tools
Over 100 analysis softwares supported for data analysis.
PyOmics
A language for faster data analysis and visualization.
Projects
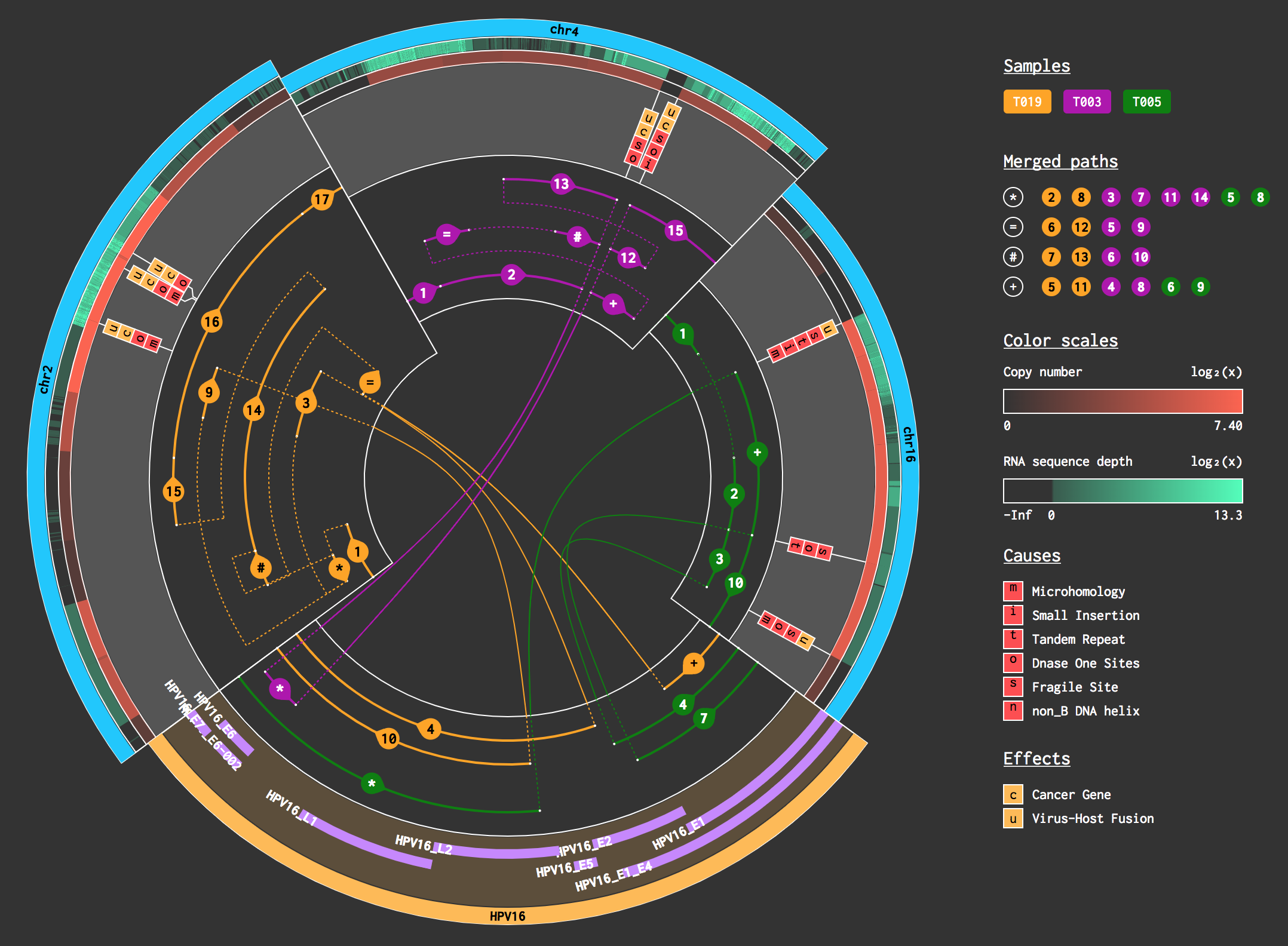
DOVIRUS
an integrated Oncovirus analysis platform
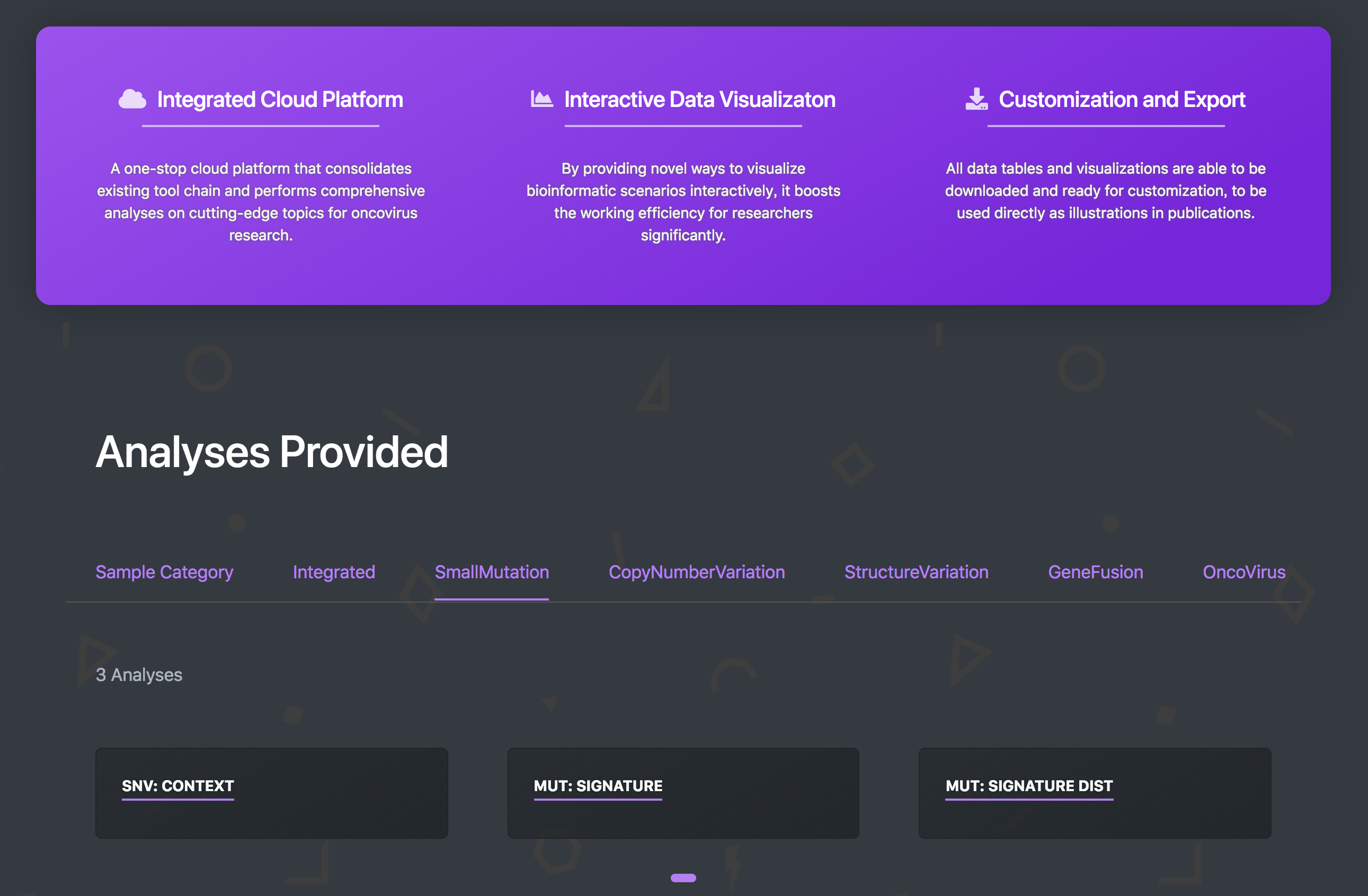
Oviz-Bio
a platform for cancer genomics data visualization
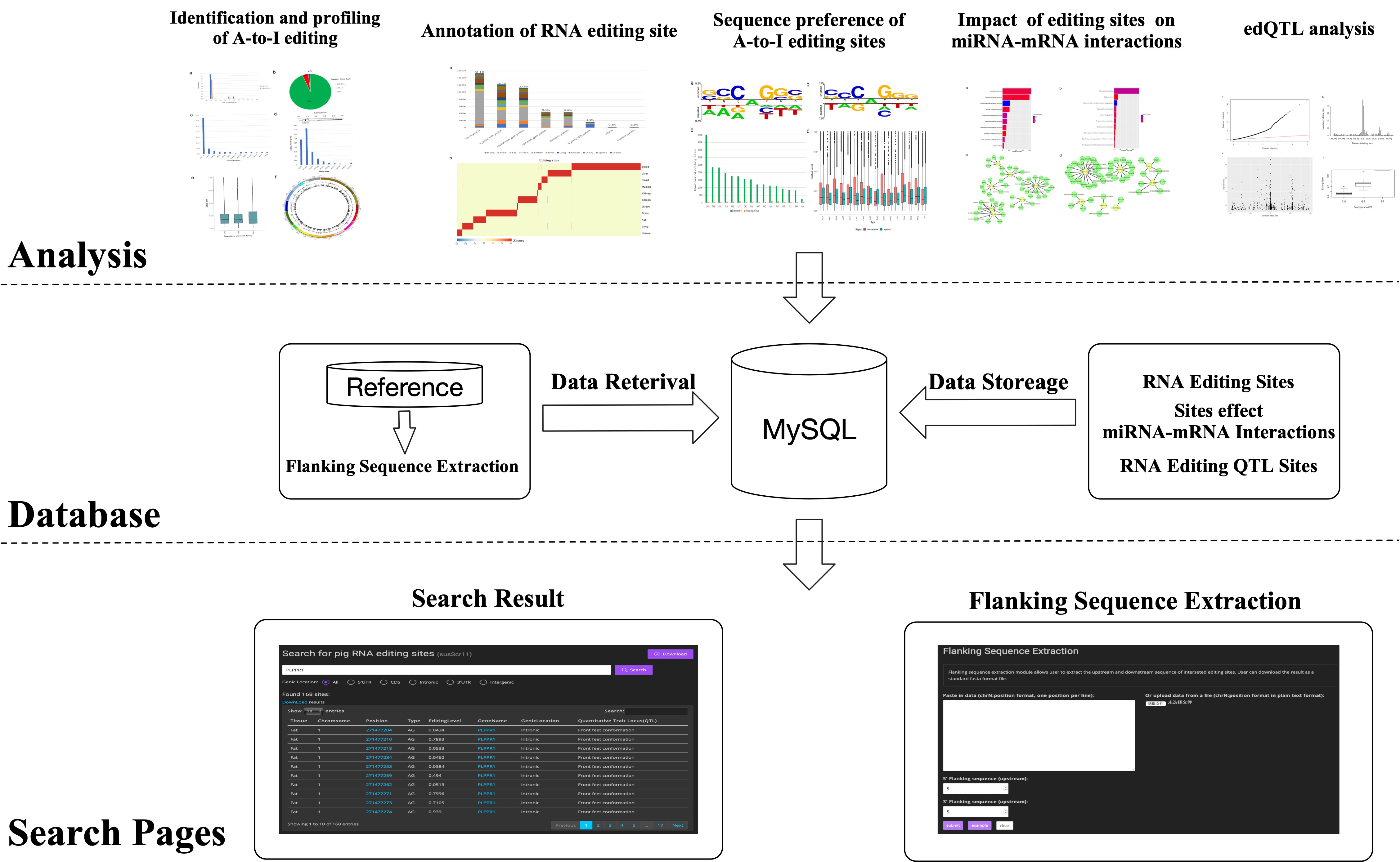
MIRIA
a webserver for statistical, visual and meta-analysis of RNA editing data in mammals
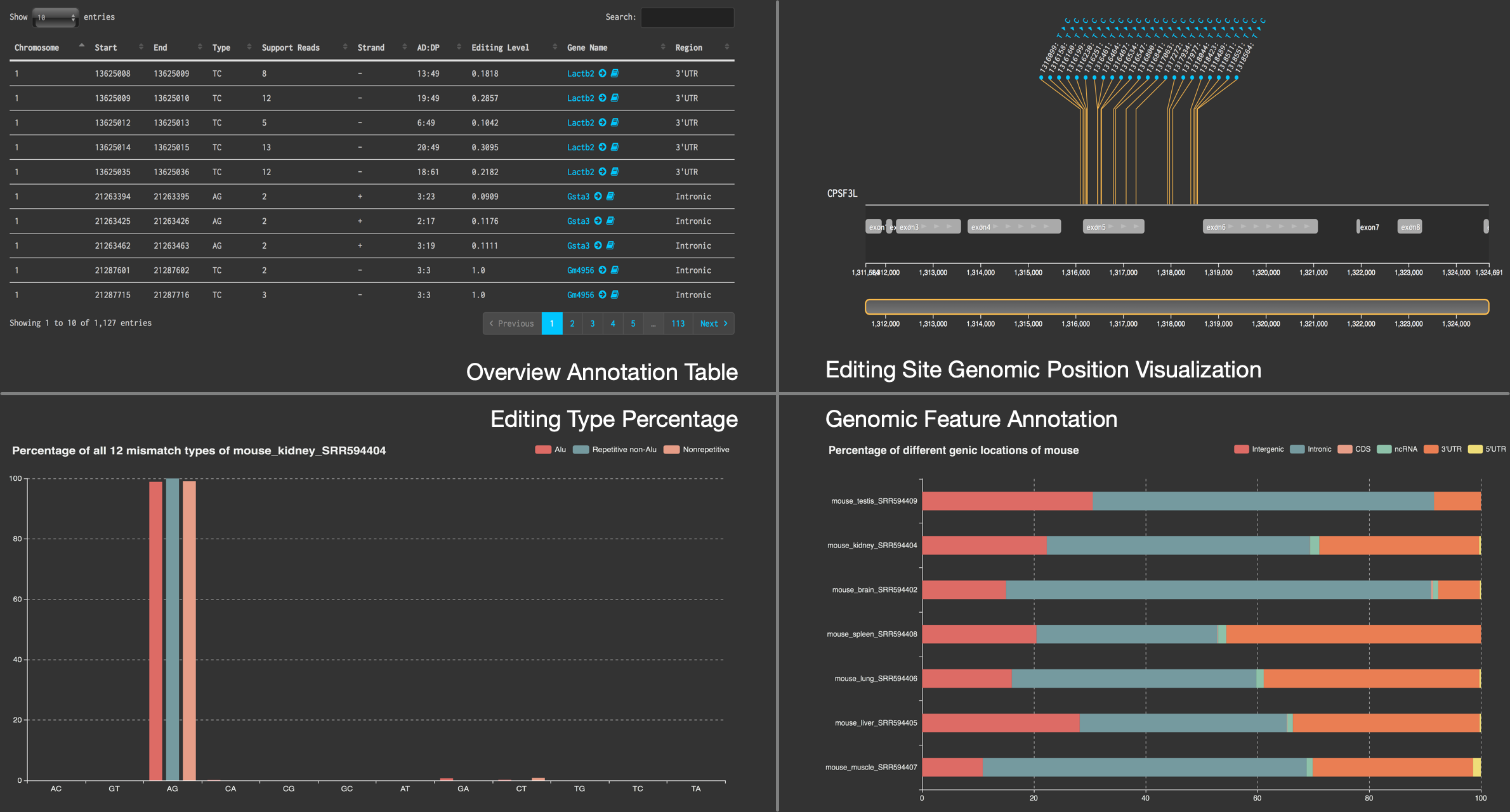
PRESDB
the DataBase of Pig RNA Editing Sites
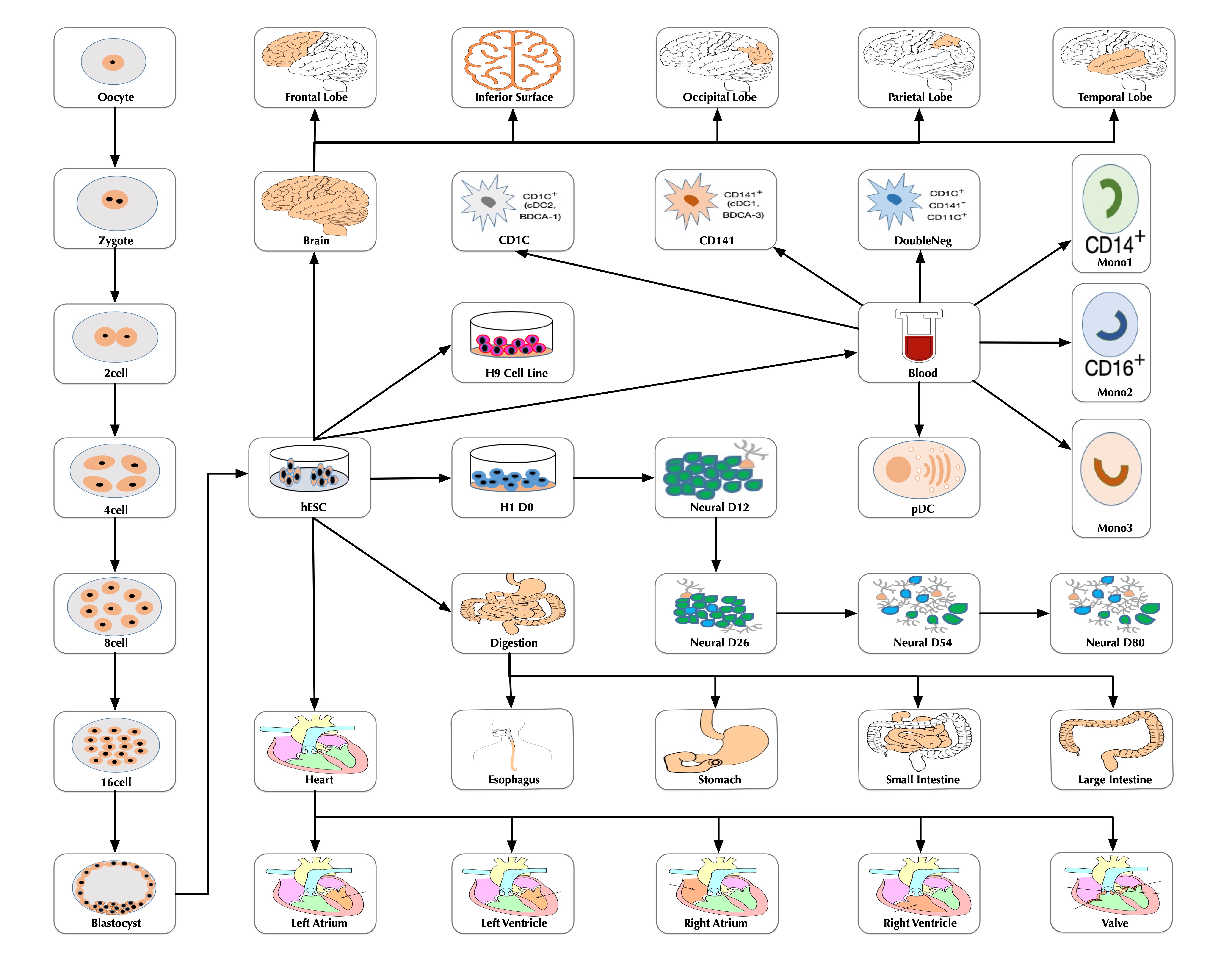
SCDevDB
A Database for Insights Into Single-Cell Gene Expression Profiles During Human Developmental Processes
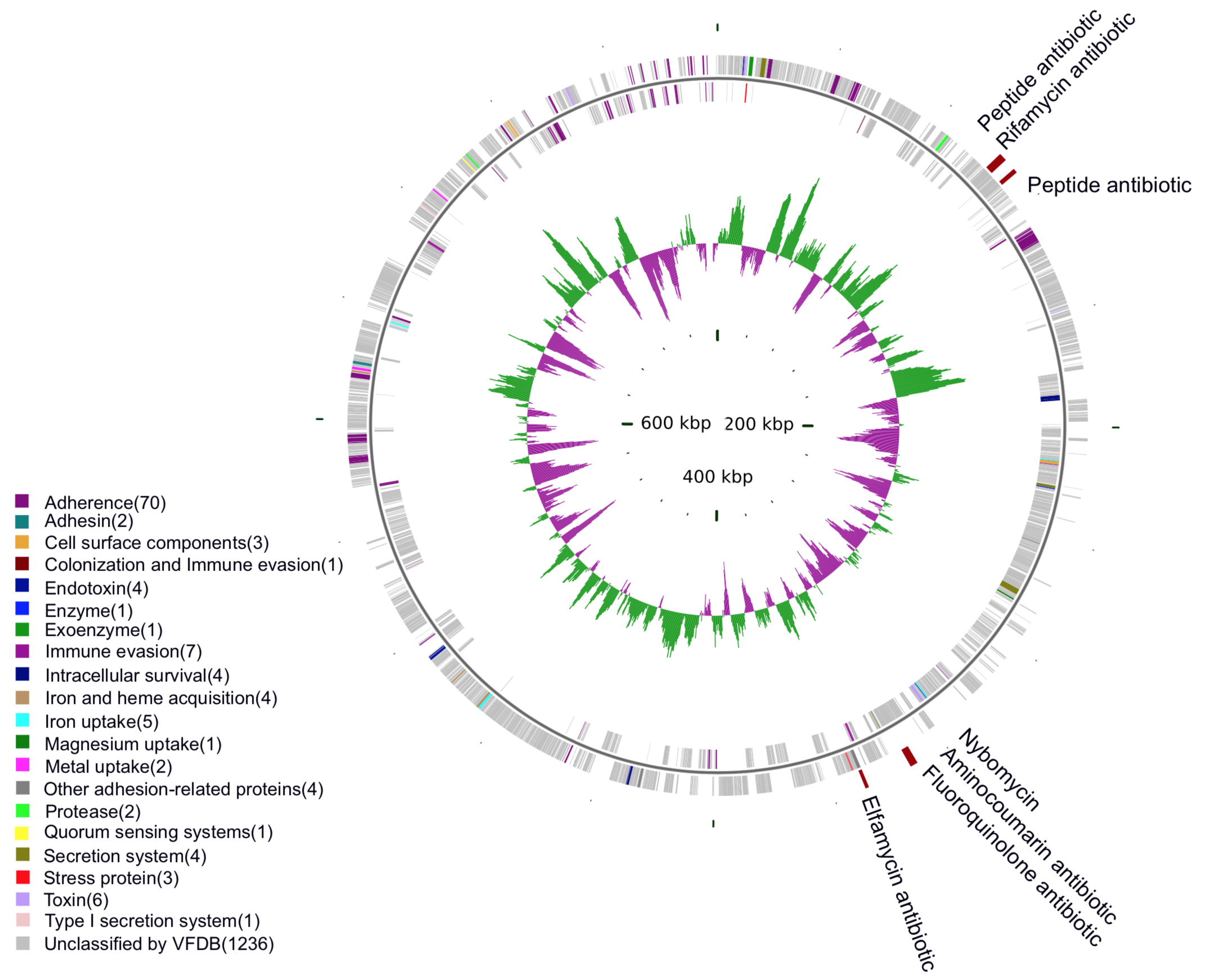
RMGC
the Respiratory Microbial Gene Catalogue
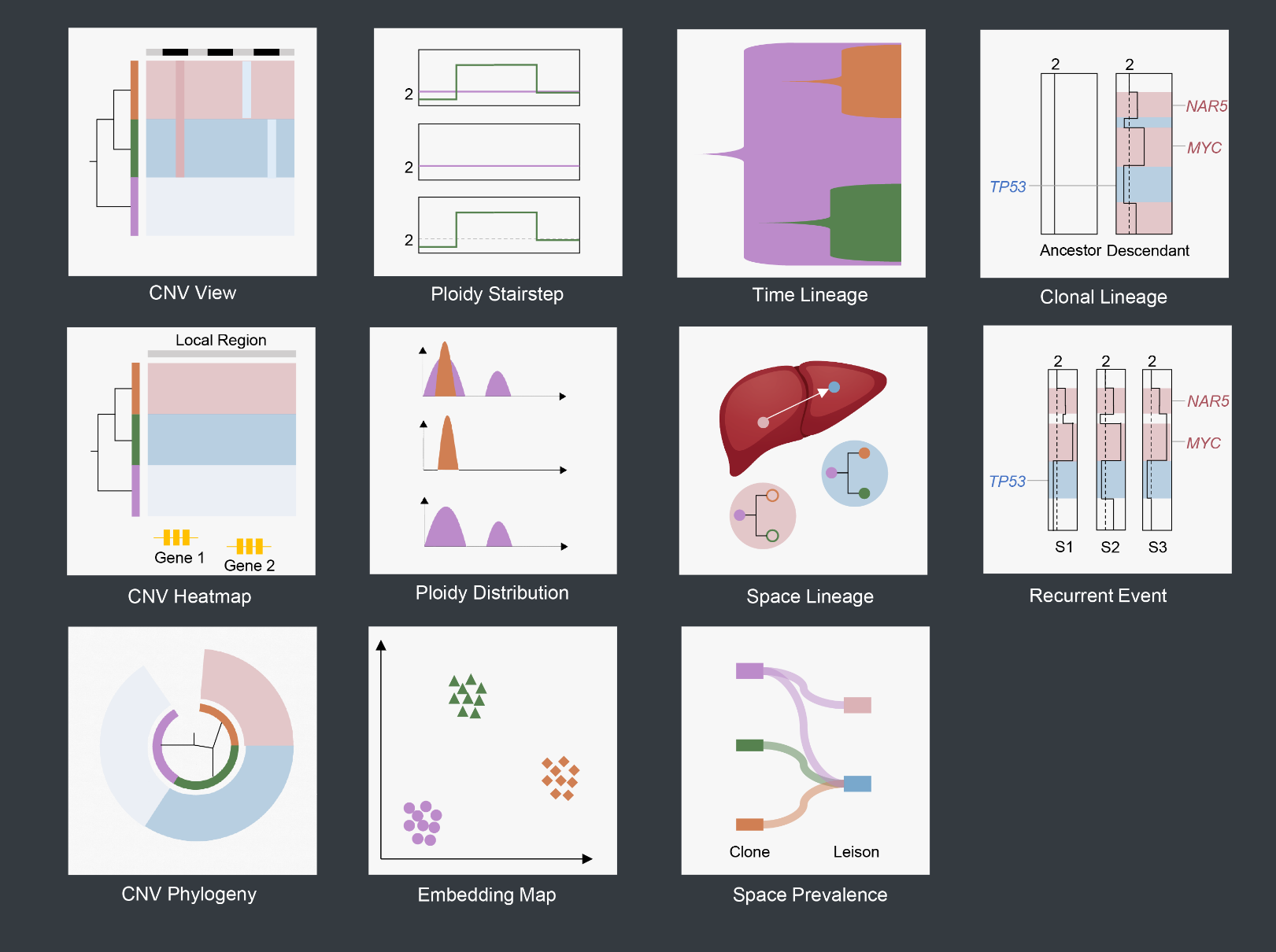
scSVAS
Platform for Single Cell DNA Visualization
MAM
a prediction tool which is developed basing on deep learning algorithms

TIMEDB
Tumor Immune MicroEnvironment cell composition DataBase

STR Profiling System
a unified STR profiling system
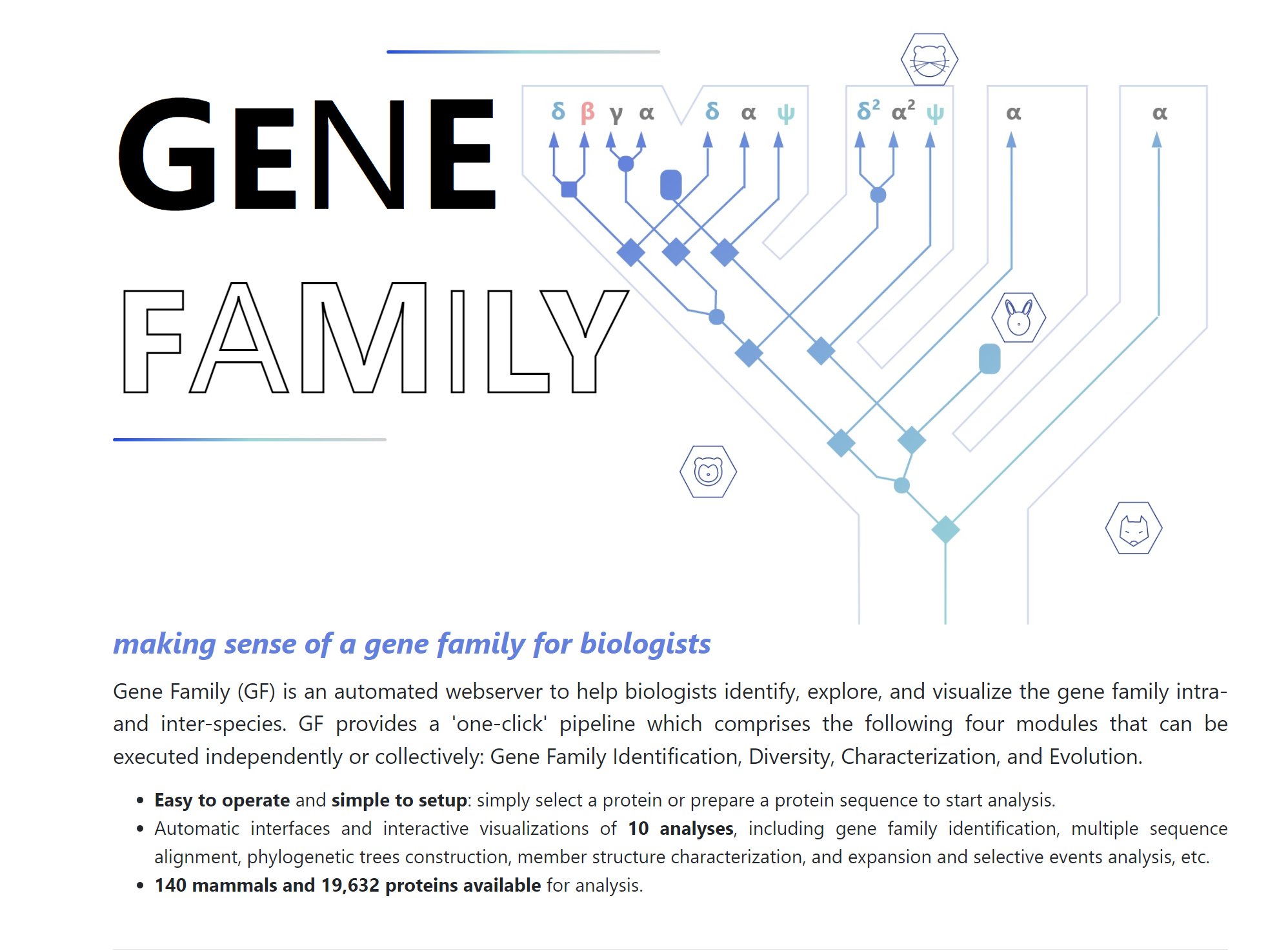
Gene Family
webserver to help biologists identify, explore, and visualize the gene family intra- and inter-species

STOMIX
Spatial transcriptomics analysis platform